Discover recent advancements in antiviral therapy, including selenium-based drugs and promising compounds targeting viral replication.
Advancements In Anti-viral Therapy:
Viruses, being intracellular parasites, hijack host cell machinery for replication and survival, making effective antiviral therapies crucial for combating viral infections, furthermore, arising sicknesses that represent a critical danger to human and creature wellbeing.
This exhaustive review provides a detailed account of recent advancements in viral therapy and antiviral drug discovery, including the development of groundbreaking selenium-based drugs that target HIV reverse transcriptase, a vital enzyme in viral replication.
Moreover, the review sheds light on the identification of Schiff base derivatives as promising antiviral agents, demonstrating potent activity against various viral strains. The review also explores the classification of viruses, the mechanisms of viral disease, and the current state of antiviral therapy, including the challenges and limitations of existing treatments.
Researchers have also identified anti-viral agents, including benzothiazoles, to combat HCV and SARS-CoV-2. Peptidomimetics, modified peptides with improved stability and bioavailability, show promise.
Structure-activity relationship studies guided the plan of peptidomimetic inhibitors focusing on SARS-CoV-2 3CLPro, leading to improved activity. Crystal structures reveal induced-fitting interactions in the S2 pocket, favoring hydrophobic alkyl groups. These findings highlight the potential of benzothiazoles and peptidomimetics as anti-viral agents.
Moreover, the 4- quinoline carboxylic acid analogue C44, discovered by SAR investigations, exhibits strong antiviral activity by inhibiting human DHODH, and X-ray crystallography confirms its promise as a broad-spectrum antiviral treatment.
Finally, the review discusses cutting-edge trends in drug development, including the application of computational science and the repurposing of approved drugs, which show great potential for the development of new antiviral medicines.
This study seeks to offer a thorough overview of the present landscape of viral treatment and antiviral drug development, as well as future directions and emerging trends in this quickly growing area.
Introduction:
Viruses are little commit intracellular parasites, which by definition contain either a RNA or DNA genome encompassed by a defensive, infection coded protein coat. Infections might be seen as versatile hereditary components, most likely of cell beginning and described by a long co-development of infection and host.
For spread infections rely upon particular host cells providing the complex metabolic and biosynthetic hardware of eukaryotic or prokaryotic cells. A total infection molecule is known as a virion.
The principal capability of the virion is to convey its DNA or RNA genome into the host cell with the goal that the genome can be communicated (interpreted and deciphered) by the host cell. The viral genome, frequently with related fundamental proteins, is bundled inside a symmetric protein capsid.
The nucleic corrosive related protein, considered nucleoprotein, along with the genome, frames the nucleocapsid. In wrapped infections, the nucleocapsid is encircled by a lipid bilayer got from the changed host cell film and studded with an external layer of infection envelope glycoproteins.
Viruses are dormant external the host cell. Little infections, e.g., polio and tobacco mosaic infection, could be solidified. Infections can’t create energy. As commit intracellular parasites, during replication, they completely rely upon the confounded biochemical hardware of eukaryotic or prokaryotic cells.
The principal motivation behind an infection is to convey its genome into the host cell to permit its demeanor (record and interpretation) by the host cell.
Classification Of Virus:
Virus are grouped based on morphology, substance synthesis, and method of replication. The infections that contaminate people are right now gathered into 21 families, reflecting just a little piece of the range of the large number of various infections whose host goes stretch out from vertebrates to protozoa and from plants and parasites to microbes. Infections are ordered based on morphology, synthetic piece, and method of replication.
The infections that taint people are right now gathered into 21 families, reflecting just a little piece of the range of the huge number of various infections whose host goes reach out from vertebrates to protozoa and from plants and parasites to microscopic organisms.
Viral Disease:
Throughout recent many years, there has been mounting interest in the rising number of infections causing surprising ailment and pandemics among people, natural life and domesticated animals.
Over and over again episodes have truly extended both nearby and public assets when medical services spending in the financially evolved world has been compelled. Significantly, ability to recognize and control arising sicknesses stays restricted in more unfortunate districts where large numbers of these illnesses have their starting point.
Arising infection sicknesses are a significant danger to human and veterinary general wellbeing.
With new models happening roughly one every year, the greater part are infections beginning from a creature have. Of the many variables dependable, changes to nearby environments that annoy the harmony among microbe and chief host species is one of the significant drivers, along with expanding urbanization of humankind and changes in human way of behaving.
Many arising infections have RNA genomes and as such are equipped for quick transformation and determination of new variations notwithstanding natural changes in have numbers and accessible objective species.
Anti-Viral Therapy:
Antiviral treatment is one of the most thrilling parts of virology, since it has effectively utilized fundamental science to produce exceptionally powerful medicines for serious viral contaminations.
Antiviral treatment is one of the most thrilling parts of virology, since it has effectively utilized fundamental science to create extremely powerful medicines for serious viral diseases.
Treatment for human immunodeficiency infection (HIV) disease has shown the potential effect antivirals can have on a deadly, persistent contamination with lifesaving treatment controlled to in excess of 12 million people by 2015.
This sensational development is going to be reiterated for the treatment of hepatitis C infection (HCV) disease. The improvement of new antiviral medications is a lot of a work underway, with dynamic medication revelation programs for filo-viruses, dengue, and others.
The reasonable way to deal with drug advancement is in transition. Previously, the essential center has been upon infection targets, and this keeps on being an extremely useful methodology.
It is currently being supplemented by a more extensive arrangement of approaches, so that current procedures include: intensifies that target nonexclusive viral targets like RNA or DNA union and could be dynamic against a scope of various infections and mixtures that are coordinated against have cell exercises important for infection replication, which could target one or a range of infections .
Moreover, laid out procedures for drug revelation are presently enhanced with the utilization of huge data sets and advancing techniques in computational science. At long last, there is an expanded accentuation on the reusing of medications previously supported for human use, driven by the exorbitant time and cost of medication advancement.
Principles Of Antiviral Therapy Virus Targets:
Existing comprehension of how individual viruses replicate at the molecular level offers detailed insights into the functions of specific viral proteins.
This understanding allows for the identification of functional regions within these proteins and the visualization of structures of them Such information can be utilized for reverse pharmacology or rational drug design involving the creation of tiny molecules that either bind to active site on viral proteins or are screened for the identification of compounds that can block specific viral activities
For instance, HIV serves as a valuable case study due to extensive efforts to develop antiviral drugs targeting various viral proteins. The majority of these anti-viral drugs focus on inhibiting either of the three viral enzymes: integrase, protease, or reverse transcriptase.
There is a wide array of drugs designed to block reverse transcription, a process not found in normal cells. These drugs fall into two categories: nucleoside and non-nucleoside reverse transcriptase inhibitors (NRTIs and NNRTIs, respectively).
NRTIs are substances that become part of the newly forming DNA chain and hinder its elongation, while NNRTIs directly bind to the enzyme, impairing its function and potentially leading to its breakdown.
Another significant target for enzymatic drug intervention is the viral protease, which cleaves specific viral polypeptides to generate mature proteins. Protease inhibitors typically attach to the catalytic site on the protease molecule.
Cellular Targets:
Theoretically, cellular targets are superior to viral targets because of the property that they are less likely to develop escape mutations. However, targeting cellular molecules may disrupt vital host functions,
Which could pose challenges for treatment especially long tern but may not be a significant obstacle for infections that are acute. Historically, cellular targets were not a major focus, they are now gaining increasing attention. For instance, CCR5, a chemokine receptor, acts as a co receptor for HIV.
Certain individuals with a mutation in CCR5 are highly resistant to HIV infection without experiencing any adverse effects, suggesting that inhibitors targeting this receptor domain could be used safely for HIV treatment. Maraviroc is one such inhibitor that has proven effective.
Cyclosporin A (CsA) is another example of a drug that targets a cellular molecule, cyclophilin A, which is involved in various cellular processes necessary for HCV replication. Despite its role in HCV replication, cyclophilin A does not appear to be essential for life, as evidenced by healthy mice with knockouts of this molecule.
Viral Pathogenesis And Anti-Viral Strategy:
Recognizing the epidemiological features, pathophysiology, and transmission of of different Viruses is crucial in determining the possible effectiveness of antiviral drugs.
Viruses with short incubation periods and rapid spread are often challenging targets for antiviral treatment, as timely diagnosis and therapy initiation can be difficult.
For example, influenza has a brief incubation period of 18-72 hours, so neuraminidase inhibitors must be administered prior to the infection or shortly after symptoms arise to be effective.
However, during a pandemic outbreak, widespread administration of antiviral drugs as a short-term preventive measure could help mitigate potential infections.
On the other hand, chronic viral infections present a good target for antiviral treatment. Their gradual course allows for precise diagnosis and evaluation prior to initiation of the therapy..
Diseases like hepatitis B and hepatitis C, with millions of cases globally and long-term risks such as liver failure or cancer, present significant opportunities for treatment.
There is a delicate balance between the host’s defences and the virus in persistent infections, as evidenced by the fact that some people are able to overcome the infection after years of persistence. According to this, antiviral medication may be able to tip the scales in the host’s favour and cause the virus to go.
Additionally, combining antiviral drugs with antiviral antibodies may enhance therapeutic outcomes
Drug Resistant Viral Mutations:
Antiviral therapy is significantly hampered by drug-resistant viral mutations, which can be caused by a number of reasons, including:
- Because RNA polymerases lack physiological proofreading mechanisms and have poor fidelity, RNA viruses have a significantly higher rate of mutation than DNA viruses.
- The virus replication rate during an infection significantly impacts the production rate of mutant virions. For example, HIV-1 produces a large number of virions daily, leading to numerous mutants, while viruses like HPV replicate slowly, resulting in fewer mutant virions.
- Different drugs target diverse viral functions with varying importance for replication, and their efficacy in blocking these functions differs. Furthermore, resistance mutants differ in their potential for replication both when the medication is present and when it is not present.
- A drug’s in vivo selective pressure is determined by its ability to disrupt important virus functions as well as its pharmacodynamics, which control its concentration at viral replication sites. The selection of escape mutants is maximized when the medication concentration is high enough to select for resistant mutants but not so high as to severely impair virus replication, lowering the likelihood of resistant virus emergence.
Pharmacodynamics:
A vital component of a medication’s efficacy is how it behaves in a living organism. Many compounds that demonstrate activity in cell cultures may not prove effective when they are being tested in animals.
A drug’s pharmacodynamics depend on a number of important factors:
- Solubility of the compound.
- Absorption capability, whether orally, through injection, or intravenously.
- Activation process, whether the drug is active in its administered form or requires liver processing for activation.
- Rate of release into the bloodstream and its circulation, whether as a free molecule or bound to plasma proteins like albumin.
- Site of action, whether in the blood or specific target tissues, and the speed of entry into these tissues.
- Half-life in both blood and tissues.
- Metabolism in the liver and excretion via urine or the intestinal tract.
- Reaching therapeutic concentrations in target tissues and blood, as well as the required dosing schedule to keep these concentrations there.
To ascertain whether a candidate compound satisfies these pharmacodynamics requirements for practical application, a number of tests must be conducted.
Selenium Based Antiviral Drugs:
A comprehensive review of selenium-based drugs that target HIV reverse transcriptase (RT) inhibitors opens up new and promising therapeutic approaches for HIV infection. Nucleoside reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs) are two categories of these substances that have different modes of action but share the same objective of blocking HIV RT in order to prevent viral replication.
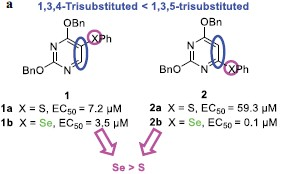
The 1,3,5-trisubstituted analogues of phenylselenenyl- substituted pyrimidines, such as compound 2b, show greater anti-HIV-1 activity. Furthermore, compounds 1a and 1b demonstrate significant activity in phenoxy substitutions.
In recent study, the powerful anti-HIV activity of oxaselenolane nucleosides, which have selenium substitution at the 3′-CH2 position, is demonstrated by the cytosine analogues 7c (EC50 = 2.7 μM) and 5-fluorocytosine analogue 7e (EC50 = 0.73 μM). The efficacy is higher in the (-)-enantiomers and β- substituted variants. These substances have excellent safety profiles across a variety of cell lines,
suggesting that they may be novel anti-HIV agents.Similar to this, compounds 8a, 8b, 8c, and 9b show encouraging efficacy, however toxicity concerns exist. Selenium-modified 5′-Homo- 4′-Se-d4Ns also exhibit anti-HIV activity. SAR research emphasizes that amino substitutions are more crucial for enhancing chemical activity than hydroxyl and hydrogen substitutions.
The derivatives of 1,2,3-selenadiazole thioacetanilides (STA) have been shown to be effective NNRTIs against HIV-1. Among these, compound 10f has the highest potency (EC50 = 2.45 ± 1.08 μM). According to SAR studies, benzene rings that are ortho- or para-substituted increase activity.
Derivatives 10h, 10i, 10l, and 10k show significant anti-HIV activity and emphasize the potential of selenium-based NNRTIs in HIV research, despite their relative ineffectiveness.
A promising direction in the development of antiretroviral drugs has been highlighted by the identification of 2,2′-diselenobisbenzamides (DISeBAs) as novel HIV inhibitors. These substances, which were discovered by Sancineto et al., have strong anti-HIV-1 and anti-HIV-2 activity in MT-4 cells and inhibit NCp7 and interfere with the processing of the gag precursor.
Antiviral activity is enhanced by alkyl or long-chain carboxyl substitutions, while ester groups are not preferred over amide connections, according to SAR studies. However, toxicity increases with esterification of carboxyl groups, and amino cyclization diminishes activity. Asquith et al. presented 1,2,3- thiaselenazoles as possible HIV NCp7 inhibitors in a recent study, showcasing their effectiveness against HIV and the Feline Immunodeficiency Virus (FIV).
Compounds 15b and 16f had noteworthy anti-FIV action, highlighting the superiority of selenium over sulphur substitution in terms of increased efficacy. The 1,2,3-thiaselenazole ring is essential for action, as demonstrated by SAR investigations, which provide important information for developing new anti-HIV treatments.
The discovery of ebselen as an HIV-1 capsid CTD inhibitor has generated interest in its analogs. Through structural variations, such as replacement with carbon, sulphur, or oxygen, or through modifications to the phenyl ring, SAR investigations clarified the role that the selenium atom plays in the activity of ebselen.
Among the analogs, 2-pyridyl-ebselen and ebselen oxide demonstrated encouraging potency and were found to have cell viability similar to ebselen, suggesting possible ways to overcome drug resistance. These results demonstrate the importance of derivatives based on selenium in the continuous development of anti-HIV therapies.
Various analogs have been developed as a result of the invention of ritonavir, an innovative HIV protease inhibitor. Selenazole- substituted ritonavir (20a) was synthesized by Qiao et al. (2018), who showed similar binding to ritonavir with increased anti-HIV activity. Through improved interactions with important residues in HIV-1 protease, a further alteration to 20b that substituted isopropyl for phenyl improved binding affinity and anti-HIV efficacy.
Schiff Base In Antiviral Drug Discovery And Design
To form the nitrogen cognate known as Schiff base, imide or azomethine groups are substituted for the carbon-oxygen double bond (C = O) group. Schiff bases are commonly used carbon-based compounds.
Their biological effects include lowering body temperature, treat viral infections, reduce inflammation, restrict cell proliferation, treat malaria and have fungicidal and bactericidal properties.
Imine or azomethine groups can be found in a wide range of compounds, both natural and man-made. Imine groups are required for biological actions in these compounds. A Schiff base ligand was synthesized by combining 5-nitropyridine-2-amine and 4-hydroxy-3-methoxy benzaldehyde in a 2:1 stoichiometric ratio (2HL:M).Isatin and its Schiff base derivatives:
Isatin (1H-indole-2,3-dione) is a heterocyclic moiety that has been shown to suppress certain viral infections. Antiviral medicines having isatin nuclei include Toceranib, Semaxanib and Sunitinib.
Pandeya et al. created a norfloxacin-isatin mannich basis to assess its anti-HIV activity. Compounds 1a and 1b demonstrated protection levels ranging from 70-95% and high efficacy, with EC50 values of 11.3 and 13.9 μg/ml. Substituting electron drawing group at fifth position and trimethoprim at the third position resulted in the highest potency compared to other substituents.
In a separate series, by using the MTT 3-(4,5-dimethhyl thiazol-2-yl) assay 3 (N)-sulphadoxine of Isatin derivatives (2a-c) were investigated for HIV inhibition.
A drug with a piperidinomethyl group (2a; YP- 04) demonstrated up to 12% activity against HIV (III B) strain and 16% activity against HIV-2 (ROD) strain at an EC50 > 2 μg/ml.
SAR analysis revealed that methyl-substituted isatin had lower activity relative to unsubstituted isatin derivatives, while 3-methyl morpholine substitution at position-1 of isatin had higher potency. Two drug moieties, pyrimethamine (2b; YP-0) and sulphadoxine, provided 13% protection against HIV-2 (ROD) at an EC50 of above 2 μg/m.
Benzothiazoles As Potential Anti-Viral Agents
Anti-Hcv (Hepatitis-C Virus) Agents
Hepatitis C virus (HCV) infection is the primary cause of liver failure and frequently results in hepatocellular carcinoma, so it is a serious worldwide health concern. Approximately 150 million individuals globally suffer from HCV.
here are seven main genotypes of this single-stranded RNA virus, which belongs to the Flaviviridae family. To find inhibitors that target the viral serine protease and RNA-dependent RNA polymerase (RNA replicase), extensive high-throughput antiviral drug discovery screens were conducted.
Hepatitis A and B vaccinations are readily available, and Direct acting anti-viral drugs have been demonstrated efficacy in treating HCV infection. Yet, problems persist because of the emergence of resistance, inadequate pharmacokinetic characteristics, and expensive treatment expenses.
Therefore, there is an urgent need to develop new anti-HCV drugs. Notably, Numerous analogues of benzothiazole have signified encouraging anti-hepatitis C virus properties.
HCV NS3 Helicase Inhibitors:
The HCV replication depends on the non-structural protein 3 (NS3), which performs helicase and protease activities in mammalian cells. The 3000 amino acid HCV polyprotein is separated into two regions: non-structural (NS2-NS5B proteins) and structural (C-p7 proteins).
Since only the NS3-NS5B region is necessary for genome replication in cell cultures, NS3 is an important target for antiviral medicines because of its function in viral replication.
Li et al. conducted a high-throughput screen (HTS) of 827 compounds from the National Cancer Institute (NCI, US) mechanistic set and discovered that the commercial dyes thioflavin S and primuline were potent inhibitors of NS3-catalyzed DNA and RNA unwinding at micromolar concentrations.
A strong benzothiazole tetramer was discovered after further refinement. The specificity was increased by adding a carboxamide group, which produced a helicase inhibitor with an IC50 of 2.6 µM.
HCV NS5B Polymerase Inhibitors:
HCV NS5B polymerase replicates the positive-strand genomic RNA that host mammalian cells lack, so it presents a potential target for HCV therapy. Manfroni et al. used a scaffold-hopping technique to discover novel pyridobenzothiazole compounds that inhibit HCV NS5B polymerase. While these drugs shown potential polymerase inhibitory activity, their low permeability limited their projected anti-HCV effect in vitro.
Detailed biochemical studies demonstrated that pyridobenzothiazoles affect the polymerase allosterically, serving as non- competitive ribonucleotide substrate inhibitors and competitive RNA template inhibitors.
In their quest for new small molecule inhibitors of HCV replication, researchers at Merck Research Laboratory in the United States discovered a new library of carbanucleoside derivatives as lead compounds.
To enhance the pharmacokinetic characteristics and efficacy of these inhibitors, pyrimidine core structure-activity relationship (SAR) studies were carried out. It was discovered that the ideal substitute for the pyrimidine ring’s 5-position was a benzothiazole group. With favorable rodent plasma/target organ concentrations, strong replicon potency, and selectivity, the 4-methyl derivative has a better in vivo profile in rats. Moreover, more potent pyridothiazole analogues were discovered by appending a nitrogen atom to the benzene ring of a previously discovered benzothiazole inhibitor of HCV replication.
Modern Advancement And Strategies In Antiviral PeptidomimetICS:
Over 641 million infections and 6.62 million fatalities have been attributed to the COVID-19 pandemic, which is the deadliest of the twenty-first century and is caused by SARS-CoV-2.
Even though chemotherapeutics have been developed to treat viruses such as HIV and influenza, the creation of viruses that are resistant to drugs and the regular introduction of new viruses present substantial hurdles to the efficacy of current treatments.
When it comes to efficacy, safety, and tolerance, peptides which are essential for many biological processes like immune response, metabolism, and respiration offer attractive characteristics for potential therapeutic applications. However, issues like quick deterioration, poor absorption, limited dispersion, and off-target effects make it difficult to use them in therapeutic settings.
Peptidomimetics, or altered peptides with improved stability, bioavailability, and receptor- binding affinity, are being developed as a solution to these problems. Medicinal chemistry techniques such as terminal modifications, pseudopeptides, amino acid modifications, inverse-peptides, cyclization, and molecular hybridization are described in this review in order to create antiviral peptidomimetics.
Paxlovid, N-0385, and 11L are a few recent examples of effective antiviral drugs that are reviewed, demonstrating the promise of peptidomimetics as a potent strategy in antiviral medication development.
Structure Activity Relationship:
“To optimize a peptidomimetic inhibitor for SARS-CoV-2 3CLPro, we analyzed the co-crystal structure of inhibitor 10a with 3CLPro (PDB: 7DHJ). Our findings revealed that 10a binds to the active site through a covalent bond with Cys145, hydrogen bonding with His163, and occupying the S1 and S2 pockets. The S2 pocket, Which is hydrophobic and surrounded by specific residues, was identified as a target for optimization.
We aimed to introduce a hydrophobic alkyl group with suitable shape and size to occupy this pocket and enhance inhibitory activity. Additionally, we sought to introduce a π-electron group at the P2 site to form a π-π stacking interaction with His41, further improving inhibitory activity.”
Design And Synthesis Of Peptidomimetic Inhibitor:
“Design and synthesis of peptidomimetic inhibitors targeting SARS-CoV-2 3CLPro. Based on analysis of the S2 pocket, two new inhibitors (10b and 10c) with different alkyl groups at the P2 position were designed and synthesized.
Evaluation of inhibitory potency revealed that 10b (IC50 = 0.374 ± 0.006 μM) and 10c (IC50 = 0.373 ± 0.011 μM) showed improved inhibitory activity compared to 10a and the positive control. Crystal structures of 3CLPro in complex with 10b (PDB: 7DGB) and 10c (PDB: 7DGG) revealed that the aldehyde group of each inhibitor forms a covalent bond with Cys145, while the P2 group binds to the S2 pocket.
he structures showed that the flexible isobutyl and n-butyl groups of 10b and 10c, respectively, fit better in the S2 pocket, improving hydrophobic interactions, whereas the rigid alkynyl group of 10a failed to interact with His41.
The results demonstrate that a hydrophobic alkyl group at the P2 site is favored for inhibitory activity and that the S2 pocket interacts with peptidomimetic inhibitors in an induced-fitting mode, accommodating branched or long-chain alkyl groups.”
Quinoline Carboxylic Acid Analogue With Potent Antiviral Activity:
In this study, Das et al. indicate the identification and optimization of a 4-quinoline carboxylic acid analogue, C44, which shows potent antiviral activity through inhibition of human dihydroorotate dehydrogenase (DHODH).
The study addresses the issue of drug resistance in antiviral therapies that target viral proteins by focusing rather on host factors that contribute to viral replication. In order to maximize the antiviral properties of these substances, the authors carried out structure–activity relationship (SAR) studies.
They started with a high-throughput screening hit that resulted in the identification of C44.
The first SAR investigations concentrated on the right-hand aryl ring and investigated a range of substitutions on the quinoline carboxylic acid scaffold. The activity of compounds containing various alkoxy groups (C2-C4) varied, with the trifluoromethoxy group (C5) showing a notable improvement.
While halogen replacements, especially fluorine (C6), were shown to have a positive effect, other alterations, such as alkyl groups (C8–C10), often resulted in decreased activity. Diaryl ether C11, an extra aromatic ring, suggested possibility for greater optimization.
Further alterations to the left-hand ring revealed that while other groups such as bromine (C13) and nitro (C16) were less efficient, a fluorine substitution at C(7) (C12) greatly increased activity.
Analogs C22–C36 were created by further refining the diaryl ether moiety; of these, the 3,4- methylenedioxyphenoxy (C30) and fluorophenyl (C31, C32) analogues continued to exhibit strong activity. The C(3)-methyl substitution (C37) and rigid planar analogues (C38) did not yield greater outcomes.
However, investigating more varied diaryl ethers produced compound C44, which exhibited a remarkable increase in potency by combining a restricted rotation with improved hydrophobicity.
C44 was substantially more powerful than brequinar in both the influenza-WSN and vesicular stomatitis virus (VSV) assays, demonstrating greater activity in suppressing VSV replication (EC50 = 2 nM). At the end of the research, the X-ray crystal structure of C44 bound to human DHODH was determined, providing important new information about the molecular interactions underlying the protein’s strong inhibition.
A crucial hydrogen bond interaction with the inhibitor carboxylate and R136 developed the majority of the hydrophobic binding site. The bulky isopropyl group on C44 may be accommodated by the structural flexibility in the DHODH active site, which improved the binding affinity of the compound.
According to this study, C44 is an appropriate choice for broad-spectrum antiviral therapy that can get beyond viral resistance mechanisms by using host-targeted-inhibition.
Method:
There are many techniques but we search out few techniques through literature and these research studies regarding “ADVANCEMENT” IN ANTI VIRAL TREATMENT” RELEVANT TO” MEDICINAL CHEMISTRY “. The 4TH Ed Lippincott, Philadelphia , Food review international , Pubmed and other electronic databases were searched.
Total of 27 articles have been selected for full-text review. In this article included selenium based antiviral drugs all substitutional compounds which are effective for advancement of HIV treatment e.g (phenylselenenyl pyrimidines substitution, selenium substitutions , 5- fluorocytosine analogue .
Selenium-modified 5′-Homo-4′-Se-d4Ns Isatin and its Schiff base derivatives also exhibit anti-HIV activity. Also discovering the compounds and its modification that are effective for HIV treatment, Schiff base in antiviral drug discovery and design) use to treating malaria bactericidal and antifungal properties.
Modern Advancement and Strategies in Antiviral Peptidomimetics, targeted to sars cov2 ans paxlovid is effective antiviral medication, quinoline carboxylic acid analogue with potent antiviral ,activity benzothiazoles as potential anti-viral agents, is also valuable technique for hepatitis c viruse.
Visit our Home Page
Future Perspective:
The future of antiviral medicine depends on a thorough study and optimization of the structure-activity relationships (SAR) of diverse pharmacophores. In selenium-based antiviral medicines, the selenium atom is critical for controlling oxidative stress and impacting viral propagation.
Future medicines should optimize the selenium-containing moiety to improve potency, selectivity, bioavailability, and minimize toxicity. Schiff bases, which include an azomethine (C=N) group, are essential for building complexes with metal ions and interacting with viral proteins.
Future designs could incorporate other substituents around the azomethine group to improve binding affinity and specificity, such as hydroxyl, amine, or halogen, for greater potency. Benzothiazoles, which include crucial sulfur and nitrogen atoms in the thiazole ring, could be modified with substituents on the benzene ring to improve antiviral properties.
Future developments could incorporate electron-donating or withdrawing groups to increase pharmacokinetic and pharmacodynamic features, as well as combining benzothiazoles with other pharmacophores for greater efficacy.
Peptidomimetic inhibitors, which resemble natural peptides with amide bonds and side chains, should aim to improve stability and bioavailability by introducing non- natural amino acids, cyclic structures, or conformationally constrained patterns.
High-throughput screening and structure-based design will reveal effective inhibitors with little off-target effects. Finally, the activity of 4-quinoline carboxylic acid analogues is dependent on their quinoline ring and carboxylic acid group.
Future attempts could include altering the quinoline scaffold and carboxylic acid to improve potency, selectivity, and pharmacokinetic features, and even combining them with other antiviral pharmacophores to achieve synergistic effects. Researchers can produce more potent antiviral drugs by utilizing modern techniques such as computational modeling and rational drug design.
Conclusion:
In conclusion, the evolution of antiviral medicine reveals the complex relationships between viruses and their hosts. Significant progress has been made, particularly in HIV and hepatitis C treatments, by addressing both viral and cellular processes.
The expanding threat of new viral infections demands continuous research and innovation. Recent advances in selenium-based drugs and Schiff base derivatives represent captivating new therapeutic possibilities.
Benzothiazole derivatives have potential as anti-HCV medicines since they target important viral proteins such as NS3 helicase and NS5B polymerase, overcoming the limitations of current treatments.
Further optimisation of these molecules may enhance their efficacy and pharmacokinetic features. Peptidomimetics are a viable technique for producing stable, bioavailable antiviral medicines by increasing receptor affinity and stability. Recent achievements, such as Paxlovid, illustrate their ability to combat resistant infections.
This research concludes that peptidomimetic inhibitors with flexible hydrophobic alkyl groups at the P2 position have higher inhibitory activity against SARS-CoV-2 3CLPro because they fit better in the S2 pocket, highlighting the importance of hydrophobic interactions and induced-fit binding for effective inhibition. Das et al. discovered and optimised C44, a 4-quinoline carboxylic acid derivative that exhibits significant antiviral action by inhibiting human DHODH, suggesting a possible technique for overcoming viral resistance. C44’s considerable efficacy and structural insights point to its potential as a broad-spectrum antiviral treatment targeting host factors. Addressing drug resistance and improving drug pharmacodynamics remain crucial. As we get a better understanding of virus-host interactions, we should expect more effective therapies for viral illnesses, boosting global health security.
REFERENCES:
1. Sancineto L, Mariotti A, Bagnoli L, Marini F, Desantis J, Iraci N, Santi C, Pannecouque C, Tabarrini O. Design and synthesis of diselenobisbenzamides (DISeBAs) as nucleocapsid protein 7 (NCp7) inhibitors with anti-HIV activity. Journal of medicinal chemistry. 2015 Dec 24;58(24):9601-14.https://pubmed.ncbi.nlm.nih.gov/26613134/
2. Asquith CR, Meili T, Laitinen T, Baranovsky IV, Konstantinova LS, Poso A, Rakitin OA, Hofmann-Lehmann R. Synthesis and comparison of substituted 1, 2, 3-dithiazole and 1, 2, 3-thiaselenazole as inhibitors of the feline immunodeficiency virus (FIV) nucleocapsid protein as a model for HIV infection. Bioorganic & medicinal chemistry letters. 2019 Jul 15;29(14):1765- 8.https://www.sciencedirect.com/science/article/abs/pii/S0960894X19303026
3. Thenin-Houssier S, De Vera IM, Pedro-Rosa L, Brady A, Richard A, Konnick B, Opp S, Buffone C, Fuhrmann J, Kota S, Billack B. Ebselen, a small-molecule capsid inhibitor of HIV-1 replication. Antimicrobial agents and chemotherapy. 2016 Apr;60(4):2195- 208.https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4808204/
4. Qiao J, Zhao C, Liu J, Du Y. Design and synthesis of selenazole-substituted ritonavir analogs. Bioorganic & medicinal chemistry letters. 2018 Aug 1;28(14):2379- 81.https://pubmed.ncbi.nlm.nih.gov/29934245/
5. Shi D, Xu S, Ding D, Tang K, Zhou Y, Jiang X, Wang S, Liu X, Zhan P. Advances in drug structure-activity-relationships for the development of selenium-based compounds against HIV. Expert Opinion on Drug Discovery. 2024 Feb 1;19(2):139-46. https://pubmed.ncbi.nlm.nih.gov/37988053/
6. Caspar DLD: Design principles in virus particle construction. In Horsfall FL, Tamm I (eds): Viral and Rickettsial Infections in Man. 4th Ed. JB Lippincott, Philadelphia, 1975 .
7. Gelderblom HR. Structure and Classification of Viruses. In: Baron S, editor. Medical Microbiology. 4th edition. Galveston (TX): University of Texas Medical Branch at
Galveston;1996.Chapter41. Available from:https://www.ncbi.nlm.nih.gov/books/NBK8174
8. Mohamed T. El-Saadony, Nidal M. Zabermawi, Nehal M. Zabermawi, Maryam A. Burollus, Manal E. Shafi, Mahmoud Alagawany, Nahed Yehia, Ahmed M. Askar, Sara A. Alsafy, Ahmed E. Noreldin, Asmaa F. Khafaga, Kuldeep Dhama, Shaaban S. Elnesr, Hamada A. M. Elwan, Alessandro Di Cerbo, Khaled A. El-Tarabily & Mohamed E. Abd El-Hack. (2023) Nutritional Aspects and Health Benefits of Bioactive Plant Compounds against Infectious Diseases: A Review. Food Reviews International 39:4, pages 2138-2160.
9. Assessment of the broad-spectrum host targeting antiviral efficacy of halofuginone hydrobromide in human airway, intestinal and brain organotypic models. 2024, Antiviral Research
10. Kaushik S, Paliwal SK, Iyer MR, Patil VM. Promising Schiff bases in antiviral drug design and discovery. Medicinal Chemistry Research. 2023 Jun;32(6):1063-76.
11. Douglas D Richman, Neal Nathanson Viral pathogenesis, 271-287, 2016 https://doi.org/10.1016/B978-0-12-800964-2.00020-3
12. Richman DD, Nathanson N. Antiviral Therapy. Viral Pathogenesis. 2016:271–87. doi: 10.1016/B978-0-12-800964-2.00020-3. Epub 2016 Feb 12. PMCID: PMC7149377. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7149377/#s0010title
13. Shahnazarian V et al. Hepatitis C virus genotype 3: clinical features, current and emerging viral inhibitors, future challenges. Ann Gastroenterol 2018; 31(5): 541–551.
14. Lavanchy D. The global burden of hepatitis C. Liver Int 2009; 29: 74–81.
15. De Clercq E. The design of drugs for HIV and HCV. Nat Rev Drug Discov 2007; 6: 1001– 1018.
16. Smolders EJ et al. Pharmacokinetics, efficacy, and safety of hepatitis C virus drugs in patients with liver and/or renal impairment. Drug Saf 2016; 39: 589–611.
17. Raney KD et al. Hepatitis C virus non-structural protein 3 (HCV NS3): a multifunctional antiviral target. J Biol Chem 2010; 285: 22725–22731.
18. Parasrampuria DA. Delicate dance intreating hepatitis C infections and overcoming resistance. In Gupta SP eds. The Viral Proteases and Their Inhibitors, 1st edn. Cambridge, Massachusetts, United States: Academic Press, 2017.
19. Li K et al. Optimization of potent hepatitis C virus NS3 helicase inhibitors isolated from the yellow dyes thioflavine S and primuline. J Med Chem 2012; 55: 3319–3330.
20. Wei Y et al. Discovery of novel hepatitis C Virus NS5B polymerase inhibitors by combining random forest, multiple e-pharmacophore modeling and docking. PLoS One 2016; 11: e0148181.
21. Manfroni G et al. Pyridobenzothiazole derivatives as new chemotype targeting the HCV NS5B polymerase. Bioorg Med Chem 2012; 20: 866–876.
22. Kwong CD et al. Novel substituted pyrimidines as HCV replication (replicase) inhibitors. Bioorg Med Chem Lett 2012; 22: 1160–1164.
23. Arasappan A et al. 5-Benzothiazole substituted pyrimidine derivatives as HCV replication (replicase) inhibitors. Bioorg Med Chem Lett 2012; 22: 3229–3234.
24. Girijavallabhan VM et al. Synthesis and SAR of pyridothiazole substituted pyrimidine derived HCV replication inhibitors. Bioorg Med Chem Lett 2012; 22: 5652–5657.
25. Ding D, Xu S, da Silva-Júnior EF, Liu X, Zhan P. Medicinal chemistry insights into antiviral peptidomimetics. Drug Discovery Today. 2023 Mar 1;28(3):103468.
26. Wang H, Pei R, Li X, Deng W, Xing S, Zhang Y, Zhang C, He S, Sun H, Xiao S, Xiong J. The structure-based design of peptidomimetic inhibitors against SARS-CoV-2 3C like protease as Potent anti-viral drug candidate. European Journal of Medicinal Chemistry. 2022 Aug 5;238:114458.
27. Das P, Deng X, Zhang L, Roth MG, Fontoura BM, Phillips MA, De Brabander JK. SAR- based optimization of a 4-quinoline carboxylic acid analogue with potent antiviral activity. ACS medicinal chemistry letters. 2013 Jun 13;4(6):517-21.